Genetic Methods of Species Identification
DNA Barcoding

DNA Barcoding is not about product barcodes - it is a metaphor! But, it involves the idea that each species might have a unique DNA sequence which allows you to identify it from all other species.
As with product barcodes, there needs to be a standard way of generating the code and relating it to the identity. With products there is an international organisation for assigning codes to manufacturers. For living organisms, the codes are effectively assigned by evolution.
Let's begin with a region of DNA sequence in a species:
CCTAGGAGATGACCAAATCTATAATGTCATCGTCACAGCCCATGCATTCGTAATAATTTTCTTTATAGTA
If this region of DNA is not critically important to the survival of the organism then mutations might occur which are not purged. That is, the species might have individuals carrying slight variants of the sequence.
1. CCTAGGAGATGACCAAATCTATAATGTCATCGTCACAGCCCATGCATTCGTAATAATTTTCTTTATAGTA 2. CCTAGGAGAAGACCAAATCTATAATGTCATCGTCACAGCCCATGCATTCGTAATAATTTTCTTTATAGTA 3. CCTAGGAGATGACCAAATCTATAATGTCATCGTCACAGCCCATCCATTCGTAATAATTTTCTTTATAGTA
These are polymorphisms ("many forms") and most species have polymorphisms at many places in their genome. Polymorphisms of the type shown here are single nucleotide polymorphisms, also known as SNPs ("snips").
If a species exists for a long time, or if the species population size gets small, some of the SNP variants will become extinct, that is no longer occur in the population. This can be due simply to random events. When a gamete is produced it may be just chance which determines whether it contains one SNP variant or another. Imagine betting on the toss of a coin. Your money goes up and down depending on the run of heads and tails, but you might just lose all of it. Once you have run out of money the game is over. The same for the SNP variant - it can become extinct due to chance. Of course, if the SNP variant has an effect on the organism then it will become common or go extinct even faster.
So, we have two opposing processes occurring within a species: the gain of variation by mutation, and the loss of variation by the extinction of SNP variants.
When speciation occurs two new species arise from their ancestor species. Each of these daughter species will inherit some, or even all, of the SNPs of their ancestor. As time passes however, the processes described above will result in the gradual loss of the ancient SNPs. Those sites which once were variable will lose that variation in the two species, and each species may now have a different nucleotide. This is called a fixed difference between the species. At other sites, there may still be ancient SNP variation. Within each species new SNPs will also arise. Overall, as time passes genetic differences accumulate between species while species retain variation, even some of the ancient variation inherited from their ancestor.
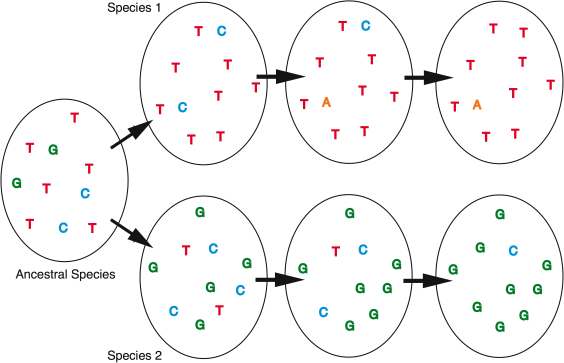
In the figure above, we see an example at one site in a gene region. The many letters represent the individuals in the species. When the ancestral species becomes two daughter species, each of them shares genetic variation with one another and with the ancestor. As time passes, the old variation is lost and new variation appears. After a while the two daughter species are noticably different at this site. And so it goes through many other sites in the gene. It is these fixed genetic differences between species which provide the DNA Barcode.
Species Identification by Barcoding
The DNA Barcoding Initiative is an international collaboration of scientists at universities and museums around the world. They are seeking a fast, reliable method for identifying large numbers of specimens, even when only fragments are available.
The Barcode of Life approach uses:
- a standard gene region so that comparisons can be made across a wide range of organisms,
- a standard set of laboratory techniques for determining the DNA sequence,
- a standard protocol (set of rules) for making an identification given a DNA sequence from an unknown specimen,
- reference sequences taken from expertly identified specimens, with good records kept of the reference specimens, and
- several reference sequences per species to provide a representative sample of the genetic variation in each species.
The standard gene region used in animals is the COI (cytochrome oxidase I, sometimes called COXI) region in the mitochondrial genome. Several different gene regions are still being tested for use in plants.
The standard protocol for making an identification is first to construct a sequence alignment between the sequence from the unknown specimen and the reference sequences. Then the similarity, or difference, between unknown and each of the reference sequences is determined. The unknown is then identified as being a member of the species from which it is most similar, provided that the difference is less than some minimum. This minimum is to protect against making an incorrect identification when the correct species is not represented among the reference sequences.
The simplest measure of similarity or difference is to determine what percentage of bases are the same or different between two sequences. Look at ostrich and emu in the following sequence alignment. How long is the alignment? How many sites are different between the two birds? What percentage of sites are different?

The alignment is 48 bases long and the two bird species differ at 6 sites, so they are 6/48 or 12.5% different or 87.5% similar.
Remember that these differences have evolved since the common ancestor of ostrich and emu. Scientists have to use a more complicated method because it may have happened that two changes have occurred at the same site, in ancestors of the emu say, or the same change could have occurred in ancestors of both birds.
That's it. Barcoding finds the sequence which is most like your unknown, and if it is not too different, then that is what species your unknown specimen belongs to.
Using DNA Barcoding
To make an identification using DNA Barcoding, go to the Barcode of Life Web site and click on the button to take you to the Identification engine.

Copy a region of the COI gene and paste it in the search window. Shortly after you click the Submit button, you will get a result similar to the following.
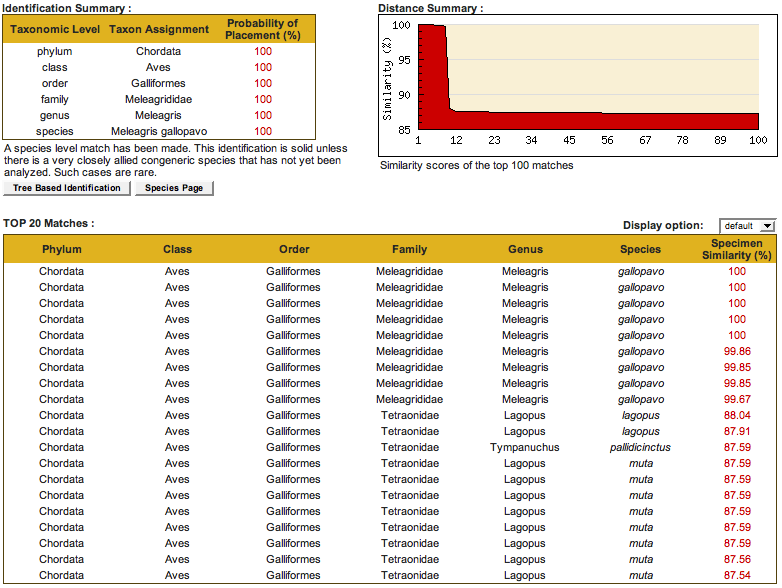
In these tables you can see that there were several reference sequences which were 100% similar to the unknown. All of these identical sequences came from the same species. There also were several sequences from the same species which were slightly different. Overall there were nine reference sequences from the most similar species (99 - 100%) before the next most similar was encountered, with a much lower level of sequence similarity (88%).
We can feel very confident that this unknown sequence is from Meleagris gallopavo.
© The University of Auckland 2020 | disclaimer